目前有研究发现,肿瘤细胞可以通过组蛋白修饰、DNA 甲基化和染色质结构改变等协同作用,通过表观调控实现免疫逃逸。前期研究发现动态可逆的 RNA m6A 甲基化与肿瘤发生和转移之间存在密切联系。考虑到 m6A 甲基化的可逆性,研究团队推测肿瘤会通过调控 m6A 甲基化过程来改变肿瘤微环境。然而,肿瘤自身的(Tumor intrinsic) RNA 表观转录组是否参与免疫逃逸仍是未知的。
2021 年 4 月 27 日,清华大学徐萌团队联合中科院北京基因组研究所韩大力团队,中科院上海药物研究所杨财广团队共同在 Cell 子刊 Cell Metabolism 发表题为 Tumors exploit FTO-mediated regulation of glycolytic metabolism to evade immune surveillance 的研究论文。
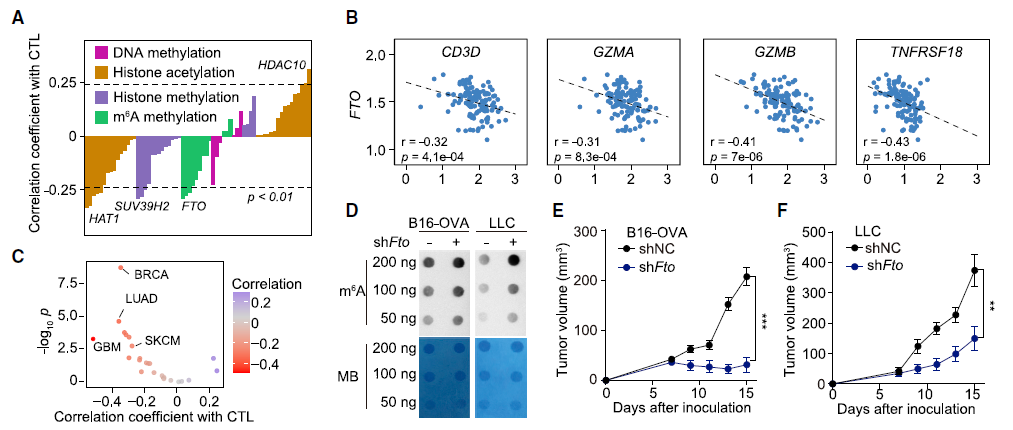
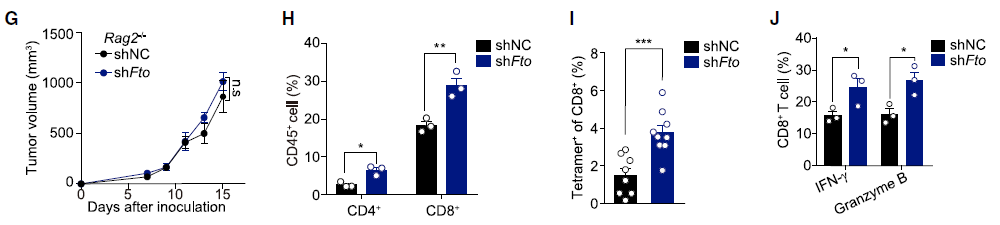
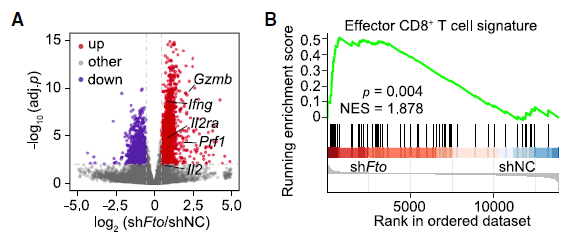
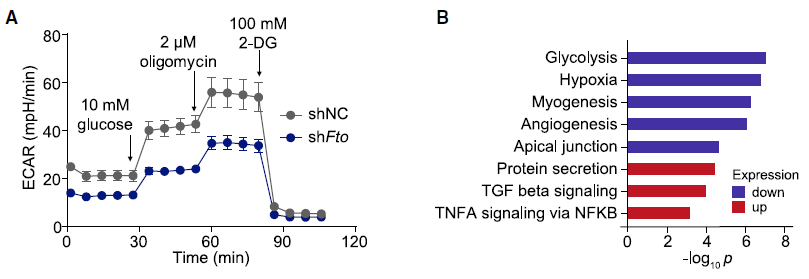
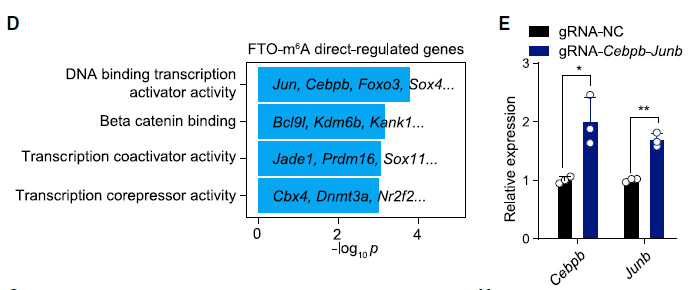
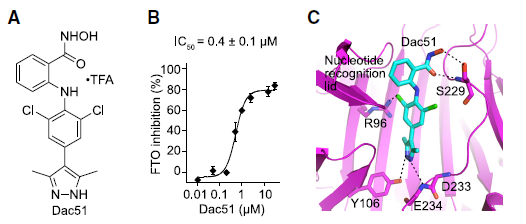
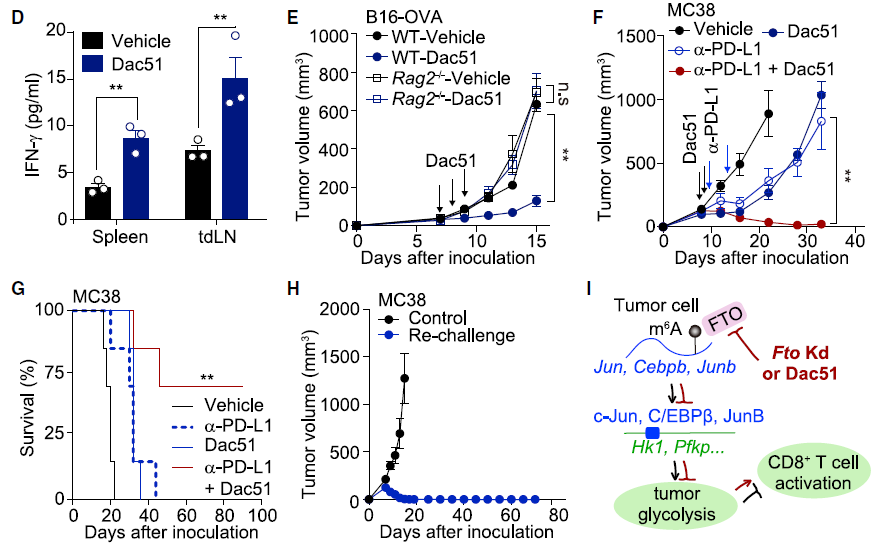
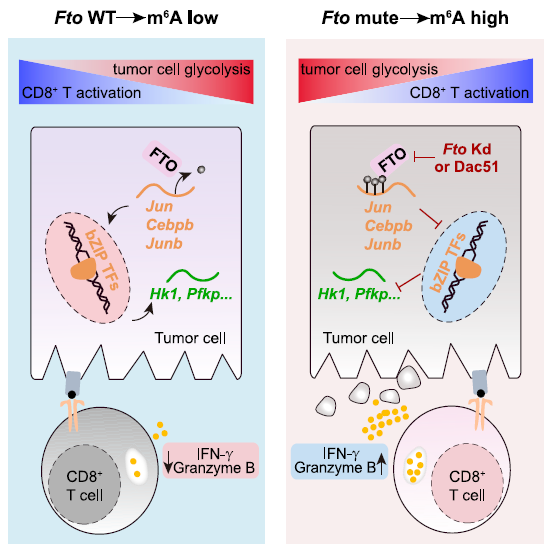
肿瘤细胞的 FTO 缺失通过增强浸润 T 细胞来抑制肿瘤生长研究人员首先分析了 TCGA 数据库的黑色素瘤(SKCM)数据,利用五个基因(CD8A, CD8B, GZMA, GZMB, and PRF1)定义了 cytotoxic T lymphocyte (CTL) score,反映肿瘤浸润 CD8+ T 细胞的功能。研究发现在表观转录组调控因子中,RNA 去甲基化酶 FTO 与 CTL score 呈现负相关。为了检测肿瘤细胞的 FTO 表达是否影响 T 细胞介导的抗肿瘤功能,研究人员在 B16-OVA 黑色素瘤细胞和肺癌细胞系 LLC 中对 FTO 进行敲降。研究人员将 B16-OVA 和 LLC 接种到小鼠体内,发现 Fto-Kd 细胞产生的肿瘤生长速度明显减慢。为进一步评估适应性免疫应答在抑制肿瘤生长中发挥的作用,研究人员将 Fto-Kd 肿瘤细胞接种在免疫缺陷 Rag2-/- 小鼠中,发现对照细胞和 Fto-Kd 的肿瘤体积相同,表明 Fto-Kd 介导的抑制肿瘤与适应性免疫应答相关。研究人员通过流式细胞术分析了肿瘤浸润的骨髓衍生细胞(单核细胞,巨噬细胞和树突细胞)和 T 细胞,发现在 Fto-Kd B16-OVA 和 LCC 肿瘤中 CD4 + 和 CD8+ T 细胞浸润明显增加。测定 IFN-γ 和 granzyme B 的产生来评估肿瘤浸润性 CD8+ T 细胞的功能,发现 Fto-Kd 肿瘤细胞 IFN-γ 和 granzyme B 阳性的 T 细胞增多。这些结果表明在肿瘤中 FTO 的缺失导致 CD8+ T 细胞浸润和其介导的细胞毒性增加。肿瘤固有 FTO 限制 CD8+ T 细胞的激活和效应状态为检测肿瘤固有 FTO 对 T 细胞的影响,研究人员对 B16-OVA 细胞和 抗原特异性 CD8+T 细胞进行共培养,随后通过流式对 CD8+ T 细胞进行分选并进行 RNA-seq。转录组分析显示,FTO 敲降导致 CD8+ T 细胞发生显著改变:1420 个基因上调和 1101 个基因下调。其中细胞因子和细胞毒性分子(IL-2, IFN-γ 和 granzyme B)的表达增加。通过对 CD8+ T 细胞效应特征基因进行基因集富集分析发现,Fto-Kd 导致 T 细胞激活并且增加了 T 细胞杀伤效果。这些结果表明肿瘤固有的 FTO 功能作为抑制因子,限制了 T 细胞的激活和效应状态。肿瘤通常利用糖酵解来促进自身快速生长,同时肿瘤细胞消耗葡萄糖会限制 T 细胞代谢,通过直接抑制其效应功能,从而促进肿瘤恶化。为检测 FTO 是否通过改变糖酵解从而限制 T 细胞的激活,研究人员测定了肿瘤细胞糖酵解的能力,发现 Fto-Kd 细胞显著降低癌细胞糖酵解的能力。研究人员通过 RNA-seq 探究 Fto-Kd 如何抑制糖酵解能力,发现在 Fto-Kd 细胞中,糖酵解途径相关基因明显下调。由于 MYC 和 bZIP 家族转录因子可以转录激活糖酵解基因,研究人员发现 Kto-Kd 细胞中 bZIP 家族转录因子 Jun,Cebpb 和 Junb 在 RNA 和蛋白水平均发生下调。为了验证转录因子与 T 细胞活化的关系,研究人员在 Fto-Kd 细胞中过表达 Junb,发现抑制了 T 细胞活化,而使用 2-DG 阻断肿瘤糖酵解后,抑制的 T 细胞活化明显被恢复。这些结果表明 Fto-Kd 通过抑制糖酵解转录因子导致 T 细胞活化增强。FTO 通过 m6A 依赖的方式调控 bZIP 转录因子为进一步探究 FTO 与转录因子之间的调控关系,研究人员对 B16-OVA 肿瘤细胞进行 M6A-seq。通过计算每个峰 m6A 比率,研究人员发现 Fto 敲降导致 m6A 甲基化整体增加。研究人员进一步挑选了 83 个 FTO-m6A 直接调控基因进行 GO 分析,发现 DNA 结合和转录激活显著富集,包括 bZIP 转录因子 Jun 和 Cebpb。为了分析这些 mRNA 中 m6A 增加是否与 FTO 直接相关,研究人员在肿瘤细胞中过表达了 CRISPR-dCas13 融合蛋白,发现在 gRNA-Cebpb-JunB 和 dCas13b-FTO 共转染时,JunB 和 C/EBPβ 及下游糖酵解基因显著上调,这些结果表明 FTO 通过去甲基化 Junb 和 Cebpb 来调控下游糖酵解基因。已有报道表明 FTO 抑制剂具有明显的抗肿瘤效果,然而目前还没有研究探究在免疫肿瘤微环境中使用 FTO 抑制剂激活 T 细胞。因此,研究人员优化了 FTO 抑制剂得到了 Dac51。体外实验表明,Dac51 对 FTO 去甲基化活性具有良好的抑制活性。此外,研究人员还测定了 FTO 和 Dac51 配合物的晶体结构,发现 Dac51 的异羟肟酸与 Ser229 之间存在额外的氢键,可能导致 Dac51 与 FTO 的结合显著增强。Dac51 处理与 Fto 敲降对糖酵解代谢的抑制水平相似。这些结果表明 Dac51 可以作为一种有效的 FTO 抑制剂,通过抑制 FTO 介导的 Jun 和 Cebpb 转录本的去甲基化,抑制肿瘤细胞的糖酵解能力。
图片来源:Cell Metabolism
Dac51 治疗增加了 T 细胞浸润,并与 PD-L1 抗体具有协同作用为了确定 Dac51 是否具有类似 Fto 敲降的抗肿瘤作用,研究人员进行体外共培养试验。在与 Dac51 预处理的 B16-OVA 肿瘤细胞共培养的 T 细胞中,发现细胞因子释放增强,细胞毒性提高。研究人员探究 Dac51 对患者来源类器官(patient-derived organoids,PDOs)的影响时发现,Dac51 处理后 c-Jun,C/EBPb 和 JunB 表达降低,编码糖酵解酶基因(LDHA 和 PFKP)同样下调,而 T 细胞激活增加。为评估 Dac51 对体内肿瘤模型的抗肿瘤作用,研究发现 Dac51 治疗可有效的抑制肿瘤在体内生长。此外,研究人员还探究了 Dac51 和免疫检查点阻断剂联合治疗的效果,发现联合治疗肿瘤生长减慢,小鼠总体生存期延长。这些结果表明,强效的 FTO 抑制剂 Dac51 可传递 T 细胞介导的抗肿瘤作用,并通过记忆 T 细胞反应防止肿瘤复发。本研究证明 Dac51 可以作为 FTO 抑制剂并阻碍 Jun,Junb 和 Cebpb 的表达,从而防止已知的破坏肿瘤内 T 细胞效应功能的代谢限制。提出了 Dac51 和 T 细胞功能增强子结合,如检查点阻断剂和其他免疫原性传统疗法,可能是协调改善适应性免疫应答的有效策略。研究人员发现 RNA 转录组可以作为免疫逃避的另一层遗传调控,这将有助于发现一类潜在的转录组免疫治疗靶点。




 您当前的位置:
您当前的位置: